
NEMLUVIO 30 mg, poudre et solvant pour solution injectable en stylo prérempli, boîte de 1 stylo prérempli de 30 mg
Dernière révision : 29/07/2025
Taux de TVA : 2.1%
Prix de vente : 2 007,43 €
Taux remboursement SS : 65%
Base remboursement SS : 2 007,43 €
Laboratoire exploitant : GALDERMA INTERNATIONAL
Source :

Dermatite atopique (DA)
Nemluvio est indiqué dans le traitement de la dermatite atopique modérée à sévère chez les patients âgés de 12 ans et plus qui nécessitent un traitement systémique.
Prurigo nodulaire (PN)
Nemluvio est indiqué dans le traitement des adultes atteints de prurigo nodulaire modéré à sévère qui nécessitent un traitement systémique.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité
Des cas d'hypersensibilité de type I, y compris d'angiœdème, ont été rapportés. En cas de réaction d'hypersensibilité systémique (immédiate ou retardée), l'administration du némolizumab doit être immédiatement interrompue et un traitement adapté instauré (voir rubrique Effets indésirables).
Aggravation de l'asthme (notamment diminution du débit expiratoire de pointe)
Dans la population de patients atteints d'un prurigo nodulaire associé à un asthme préexistant, une aggravation légère à modérée de l'asthme a été rapportée après l'initiation du némolizumab. Cet effet a été observé plus fréquemment chez des patients pesant plus de 90 kg qui avaient reçu 60 mg de némolizumab toutes les 4 semaines que chez des patients pesant moins de 90 kg qui avaient reçu 30 mg de némolizumab toutes les 4 semaines (voir rubrique Effets indésirables).
Les patients avec une exacerbation de l'asthme nécessitant une hospitalisation au cours des 12 mois précédents, les patients avec un asthme non contrôlé au cours des 3 mois précédents et les patients avec des antécédents médicaux en cours de BPCO et/ou de bronchite chronique étaient exclus des études cliniques. Aucune donnée sur l'efficacité ou la sécurité du némolizumab chez ces patients n'est disponible.
Vaccinations
Il est recommandé que les patients effectuent toutes les vaccinations appropriées à leur âge conformément aux recommandations vaccinales en vigueur avant de commencer le traitement. L'utilisation concomitante de vaccins vivants chez les patients traités par némolizumab doit être évitée. On ignore si l'administration de vaccins vivants pendant le traitement aura un impact sur la sécurité ou l'efficacité de ces vaccins. Aucune donnée n'est disponible en ce qui concerne la réponse aux vaccins non vivants.
Résumé du profil de sécurité
Les effets indésirables les plus fréquents dans la dermatite atopique et le prurigo nodulaire sont l'hypersensibilité de type I (1,1 % ; comprend l'urticaire 1,0 % et l'angiœdème 0,1 %) et les réactions au site d'injection (1,2 %) (voir rubrique Mises en garde spéciales et précautions d'emploi). D'autres effets indésirables, tels que céphalées (7,0 %), dermatite atopique (4,6 %), eczéma (3,8 %), eczéma nummulaire (3,5 %), mycoses superficielles (3,0 %) et aggravation de l'asthme (2,2 %) ont été rapportés dans le prurigo nodulaire.
Tableau des effets indésirables
Le Tableau 1 inclut tous les effets indésirables observés dans les études cliniques, présentés par classe de systèmes d'organes et par fréquence, en utilisant les catégories suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1 : Liste des effets indésirables
|
Classe de systèmes d'organes MedDRA |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Fréquent |
Infections fongiques superficielles*# |
|
Affections hématologiques et du système lymphatique |
Peu fréquent |
Éosinophilie† |
|
Affections du système immunitaire |
Fréquent |
Hypersensibilité de type I (incluant urticaire† et angiœdème*) |
|
Affections du système nerveux |
Fréquent |
Céphalées* (incluant céphalées de tension) |
|
Affections respiratoires, thoraciques et médiastinales |
Fréquent |
Aggravation de l'asthme* (incluant asthme, sibilances, diminution du débit expiratoire de pointe) |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Dermatite atopique* Eczéma*, Eczéma nummulaire* |
|
Troubles généraux et anomalies au site d'administration |
Fréquent |
Réactions au site d'injection (incluant érythème, prurit, hématome†, douleur†, irritation†, bleus* et œdème au site d'injection†) |
† Survenu dans les études sur la dermatite atopique * Survenu dans les études sur le prurigo nodulaire # Les termes Infections fongiques superficielles comprennent : Dermatophytose de la peau glabre, pied d'athlète, onychomycose, infection fongique, pityriasis versicolor, eczéma marginé de Hebra, infection cutanée fongique et infection fongique du pied
Description de certains effets indésirables
Hypersensibilité
Des réactions d'hypersensibilité de type I (réactions médiées par les IgE), incluant une légère urticaire et un léger angioœdème facial (péri-oculaire), ont été fréquemment observées chez les patients traités par némolizumab au cours des études cliniques. Ces réactions n'ont pas entraîné l'arrêt du traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Céphalée
Chez les patients atteints de prurigo nodulaire, des céphalées ont été plus fréquemment rapportées chez les patients traités par némolizumab (7,0 %) que chez les patients recevant le placebo. Des céphalées ont été plus fréquemment observées chez les femmes dans les deux groupes. Dans le groupe némolizumab, les céphalées étaient le plus souvent d'intensité légère ou modérée et n'ont pas entraîné l'arrêt du traitement.
Aggravation de l'asthme
Chez les patients atteints d'un prurigo nodulaire associé à un asthme préexistant (n = 51), 8 (15,7 %) patients ont présenté une aggravation de l'asthme après l'initiation du némolizumab, 5 d'entre eux avaient un poids corporel supérieur à 90 kg et avaient reçu 60 mg de némolizumab toutes les 4 semaines. Dans la population de patients atteints d'un prurigo nodulaire associé à un asthme préexistant, l'aggravation de l'asthme était 3 fois plus fréquente chez les patients ayant un poids corporel supérieur à 90 kg qui avaient reçu 60 mg de némolizumab toutes les 4 semaines que chez les patients ayant un poids corporel inférieur à 90 kg qui avaient reçu 30 mg de némolizumab toutes les 4 semaines.
La majorité des événements d'aggravation de l'asthme sont apparus dans les deux premiers mois de l'initiation du traitement et tous ont été rapportés comme étant d'intensité légère ou modérée. La plupart des patients ont présenté un seul événement d'aggravation de l'asthme au cours du traitement et l'événement s'est résolu avec un traitement standard par des médicaments antiasthmatiques (inhalateurs) sans corticothérapie systémique. Aucun n'a entraîné l'arrêt définitif du traitement. L'incidence des aggravations de l'asthme n'a pas augmenté avec l'exposition à long terme au némolizumab (jusqu'à la semaine 52) dans l'étude d'extension à long terme en ouvert dans le prurigo nodulaire.
Réactions eczémateuses
Chez les patients atteints de prurigo nodulaire, des réactions eczémateuses telles que la dermatite atopique, l'eczéma nummulaire ou l'eczéma ont été plus fréquemment rapportées chez les patients traités par némolizumab que chez les patients traités par placebo : dermatite atopique (4,6 %), eczéma (3,8 %) et eczéma nummulaire (3,5 %). Ces réactions eczémateuses étaient d'intensité légère ou modérée. La dermatite atopique a entraîné l'arrêt du némolizumab chez 2 (0,5 %) patients. Chez les patients âgés de plus de 65 ans, le taux de réactions eczémateuses était plus élevé.
Éosinophilie
La proportion de patients avec un taux élevé d'éosinophiles cliniquement significatif (> 700 cellules/µl) était de 10,2 % dans la population atteinte de dermatite atopique (dans la période initiale) et de 5,5 % dans la population atteinte de prurigo nodulaire. Aucune éosinophilie sévère (> 5000 cellules/µl) n'a été observée chez les patients atteints de dermatite atopique traités par némolizumab dans la période de traitement initiale. Des effets indésirables d'éosinophilie ont été rapportés chez 0,2 % des patients atteints de dermatite atopique traités par némolizumab pendant la période de traitement initiale jusqu'à la semaine 16. Tous les événements chez les patients atteints de dermatite atopique étaient d'intensité légère et n'étaient pas associés à des symptômes cliniques. Aucun évènement indésirable émergent du traitement d'éosinophilie n'a entraîné l'arrêt du traitement. À l'exception d'un cas de colite à éosinophiles chez un patient atteint de dermatite atopique associée à d'autres comorbidités atopiques, il n'y a eu aucun autre signalement de maladie à éosinophiles.
Population pédiatrique
Dermatite atopique
Adolescents (âgés de 12 à 17 ans)
La sécurité du némolizumab a été évaluée chez 176 patients pédiatriques âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère inclus dans les études ARCADIA 1 et ARCADIA 2. Le profil de sécurité du némolizumab chez ces patients jusqu'à la semaine 16 était similaire au profil de sécurité observé chez les adultes atteints de dermatite atopique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT DE COMMENCER LE TRAITEMENT :
- Effectuer toutes les vaccinations appropriées à l'âge du patient conformément aux recommandations vaccinales en vigueur.
ARRETER
LE TRAITEMENT ET SOLLICITER IMMEDIATEMENT UN AVIS MEDICAL en cas de
tout signe de réaction allergique. Les signes peuvent inclure :
- gênes respiratoires,
- gonflement du visage, de la bouche et de la langue,
- évanouissement, étourdissements, sensation d'étourdissement (dus à une faible tension artérielle),
- urticaire,
- démangeaisons,
- éruption cutanée.
Grossesse
Il existe des données limitées sur l'utilisation du némolizumab chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects en ce qui concerne la toxicité reproductive (voir rubrique Données de sécurité préclinique). Par mesure de précaution, il est préférable d'éviter l'utilisation du némolizumab pendant la grossesse.
Allaitement
Il n'existe aucune donnée sur l'excrétion du némolizumab dans le lait maternel. Chez l'humain, l'excrétion des anticorps de type IgG dans le lait a lieu au cours des premiers jours suivant la naissance, et diminue rapidement à de faibles concentrations. Par conséquent, le transfert des anticorps de type IgG aux nouveau-nés par le lait peut se produire au cours des premiers jours. Au cours de cette courte période, un risque pour les nourrissons ne peut être exclu. Par la suite, le némolizumab pourrait être utilisé pendant l'allaitement si le traitement est justifié d'un point de vue clinique.
Fertilité
Les études effectuées chez l'animal n'ont montré aucune altération de la fertilité (voir rubrique Données de sécurité préclinique).
Vaccins vivants
La sécurité et l'efficacité de l'utilisation concomitante du némolizumab avec des vaccins vivants atténués n'ont pas été étudiées. Les vaccins vivants ne doivent pas être administrés en même temps que le némolizumab (voir rubrique Mises en garde spéciales et précautions d'emploi).
Vaccins non vivants
La sécurité et l'efficacité de l'utilisation concomitante du némolizumab avec des vaccins non vivants n'ont pas été étudiées (voir rubrique Mises en garde spéciales et précautions d'emploi)
Interactions avec le cytochrome P450
Les effets du némolizumab sur la pharmacocinétique du midazolam (substrat du CYP3A4/5), de la warfarine (substrat du CYP2C9), de l'oméprazole (substrat du CYP2C19), du métoprolol (substrat du CYP2D6) et de la caféine (substrat du CYP1A2) ont été évalués dans une étude menée chez des patients atteints de DA modérée à sévère. Aucune variation cliniquement significative de l'exposition aux substrats du CYP450 n'a été observée, par rapport à la période précédant le traitement par némolizumab. Aucun ajustement posologique n'est nécessaire.
Le traitement par némolizumab doit être instauré et supervisé par des professionnels de santé expérimentés dans le diagnostic et le traitement des pathologies pour lesquelles le némolizumab est indiqué.
Posologie
Dermatite atopique (DA)
La dose recommandée est :
- Une dose initiale de 60 mg (deux injections de 30 mg), suivie de 30 mg administrés toutes les 4 semaines (1x/4 sem.)
- Après 16 semaines de traitement, pour les patients qui obtiennent une réponse clinique, la dose d'entretien recommandée est de 30 mg toutes les 8 semaines (1x/8 sem.).
Le némolizumab peut être utilisé avec ou sans corticoïdes topiques (CT). Les inhibiteurs de la calcineurine topiques (ICT) peuvent être utilisés, mais ils doivent être réservés uniquement aux zonessensibles, telles que le visage, le cou, les zones intertrigineuses et génitales. Toute utilisation de traitements topiques doit être diminuée progressivement, puis arrêtée lorsque la maladie s'est suffisamment améliorée.
L'interruption du traitement doit être envisagée chez les patients qui ne présentent aucune réponse après 16 semaines de traitement contre la dermatite atopique. Certains patients présentant initialement une réponse partielle peuvent bénéficier d'une amélioration en poursuivant le traitement après 16 semaines.
Une fois la réponse clinique obtenue, la dose d'entretien recommandée de némolizumab est de 30 mg toutes les 8 semaines.
Prurigo nodulaire (PN)
La dose recommandée pour les patients pesant moins de 90 kg est une dose initiale de 60 mg (deux injections de 30 mg), suivie de 30 mg administrés toutes les 4 semaines (1x/4 sem.).
La dose recommandée pour les patients pesant 90 kg ou plus est une dose initiale de 60 mg (deux injections de 30 mg), suivie de 60 mg administrés toutes les 4 semaines (1x/4 sem.).
L'interruption du traitement doit être envisagée chez les patients qui ne présentent aucune réponse au niveau du prurit après 16 semaines de traitement contre le prurigo nodulaire.
Dose oubliée
En cas d'oubli d'une dose, celle-ci doit être administrée dès que possible. Par la suite, l'administration doit être reprise à l'heure habituelle prévue.
Populations particulières
Personnes âgées (65 ans et plus)
Aucun ajustement posologique n'est recommandé chez les patients âgés (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique et rénale
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique ou rénale (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
Dermatite atopique
La sécurité et l'efficacité du némolizumab chez les enfants âgés de moins de 12 ans et pesant moins de 30 kg n'ont pas encore été établies. Aucune donnée n'est disponible.
Prurigo nodulaire
La sécurité et l'efficacité du némolizumab chez les enfants âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie sous-cutanée.
L'injection sous-cutanée doit être effectuée dans la partie supérieure avant de la cuisse ou dans l'abdomen en évitant la zone de 5 cm autour du nombril. L'injection dans la partie supérieure du bras ne doit être effectuée que par un soignant ou un professionnel de santé.
Pour les doses suivantes, il est recommandé d'alterner les sites d'injection pour chaque dose. Le némolizumab ne doit pas être injecté dans une peau sensible, enflammée, enflée, lésée ou présentant des hématomes, des cicatrices ou des plaies ouvertes.
Le némolizumab est destiné à être utilisé sous la supervision d'un professionnel de santé. Si le professionnel de santé juge que cela est approprié, le patient peut s'auto-injecter le némolizumab ou le soignant peut l'administrer. Avant la première injection, les patients et/ou les soignants doivent recevoir une formation adaptée sur la préparation et l'administration du némolizumab conformément aux instructions d'utilisation en fin de notice.
Durée de conservation :
Nemluvio 30 mg, poudre et solvant pour solution injectable en stylo prérempli
3 ans
Une fois les étapes de reconstitution terminées, Nemluvio doit être utilisé dans les 4 heures, conservé à température ambiante (ne dépassant pas 25° C) ou jeté.
Si nécessaire, la boîte contenant le stylo prérempli peut être sortie du réfrigérateur et conservée à température ambiante (ne dépassant pas 25 °C) pendant une période unique de 90 jours maximum. La date de sortie du réfrigérateur doit être inscrite dans l'espace prévu sur le conditionnement extérieur. Ne pas utiliser Nemluvio si la date de péremption est dépassée ou si le produit est laissé hors du réfrigérateur pendant plus de 90 jours (selon la première éventualité).
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
À conserver dans l'emballage d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'y a pas de traitement spécifique en cas de surdosage par le némolizumab. En cas de surdosage, le patient doit être surveillé afin de déceler des signes ou symptômes d'effets indésirables et un traitement symptomatique approprié doit être immédiatement instauré.
Classe pharmacothérapeutique : Autres préparations dermatologiques, médicaments utilisés dans la dermatite, à l'exclusion des corticostéroïdes, Code ATC : D11AH12
Mécanisme d'action
Le némolizumab est un anticorps monoclonal de type IgG2 humanisé qui inhibe la signalisation de l'interleukine-31 (IL-31) en se liant sélectivement au récepteur alpha de l'interleukine-31 (IL-31 RA). L'IL-31 est une cytokine naturelle impliquée dans le prurit, l'inflammation, le dysfonctionnement épidermique et la fibrose. Le némolizumab a inhibé les réponses induites par l'IL-31, notamment la libération de cytokines pro-inflammatoires et de chimiokines.
Dans les études cliniques sur la dermatite atopique, il a été observé que le némolizumab modulait l'expression des gènes liée à la physiopathologie de la dermatite atopique, avec un impact principal sur les processus du système immunitaire, en diminuant le profil inflammatoire et prolifératif de cellules immunitaires spécifiques (lymphocytes T et monocytes/macrophages) sans entraîner d'immunosuppression.
Dans les études cliniques sur le prurigo nodulaire, il a été démontré que le némolizumab modulait les processus moléculaires liés à la physiopathologie du prurigo nodulaire, avec un impact sur le prurit, l'inflammation, la différenciation épidermique et la fibrose.
Effet pharmacodynamique
Immunogénicité
Des anticorps anti-médicament (AAM) ont été très fréquemment détectés. Aucune preuve de l'impact des AAM sur la pharmacocinétique, l'efficacité ou la sécurité n'a été observée.
Efficacité et sécurité cliniques dans la dermatite atopique
Adultes et adolescents atteints de dermatite atopique
L'efficacité et la sécurité du némolizumab associé à un traitement de fond topique concomitant ont été évaluées dans deux études pivots randomisées, en double aveugle, contrôlées contre placebo (ARCADIA 1 et ARCADIA 2) dans lesquelles étaient inclus, au total, 1 728 patients âgés de 12 ans et plus atteints de dermatite atopique modérée à sévère non efficacement contrôlée par des traitements topiques. La sévérité de la maladie était définie par un score IGA (Investigator's Global Assessment [Évaluation globale de l'investigateur]) de 3 (modéré) et 4 (sévère) dans l'évaluation globale de la dermatite atopique, un score EASI (Eczema Area and Severity Index [Indice de surface et de sévérité de l'eczéma]) ≥ 16, une surface corporelle atteinte minimale (Body Surface Area, BSA) ≥ 10 % et un score PP-NRS (Peak Pruritus Numeric Rating Scale [échelle d'évaluation numérique de prurit]) ≥ 4.
Les patients dans les études ont reçu initialement des injections sous-cutanées de némolizumab 60 mg, suivies d'injections de 30 mg toutes les 4 semaines (1x/4 sem.) ou d'un placebo correspondant. Des CT et/ou ICT de puissance faible et/ou moyenne étaient administrés en concomitance dans les groupes némolizumab et placebo pendant au moins 14 jours avant l'entrée dans l'étude et ont été poursuivis pendant l'étude. En fonction de l'activité de la maladie, ces traitements concomitants pouvaient être diminués progressivement et/ou arrêtés à la discrétion de l'investigateur.
Après 16 semaines, les patients ayant obtenu un score EASI-75 ou un succès selon le score IGA ont continué dans la période d'entretien de l'étude pendant 32 semaines supplémentaires afin d'évaluer le maintien de la réponse obtenue à la semaine 16. Les patients répondeurs au némolizumab ont été randomisés à nouveau pour recevoir soit du némolizumab 30 mg toutes les 4 semaines, du némolizumab 30 mg toutes les 8 semaines soit un placebo toutes les 4 semaines (tous les groupes ont continué le traitement de fond par CT/ICT). Les patients randomisés pour recevoir le placebo au cours de la période initiale de traitement ayant obtenu la même réponse clinique à la semaine 16 ont continué à recevoir le placebo toutes les 4 semaines. Les patients non-répondeurs à la semaine 16, les patients présentant une perte de réponse clinique pendant la période d'entretien et les patients ayant terminé la période d'entretien ont eu la possibilité d'être inclus dans l'étude en ouvert (ELT
ARCADIA, ELT : Etude Long Terme) et de recevoir un traitement par némolizumab 30 mg toutes les 4 semaines pendant au maximum 200 semaines.
Critères d'évaluation
Dans les études ARCADIA 1 et ARCADIA 2, les critères d'évaluation principaux étaient les suivants :
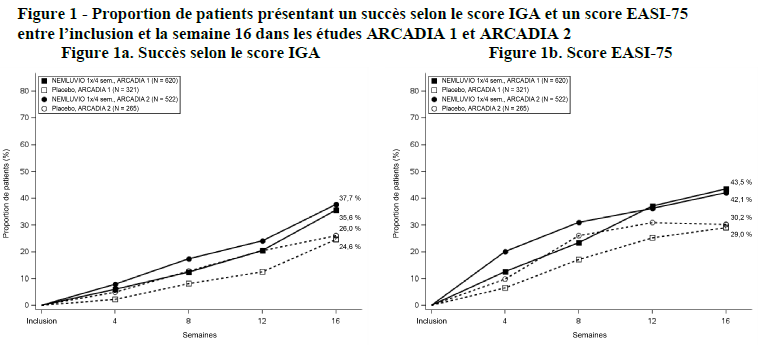
- Proportion de patients présentant un succès selon le score IGA (défini comme un score IGA de 0 [blanchi] ou 1 [presque blanchi] et une réduction ≥ 2 points par rapport à l'inclusion) à la semaine 16
- Proportion de patients présentant un score EASI-75 (amélioration ≥ 75 % du score EASI par rapport à l'inclusion) à la semaine 16
Les principaux critères d'évaluation secondaires comprenaient une amélioration du score PP-NRS ≥ 4 par rapport à l'inclusion aux semaines 1, 2, 4 et 16, un score PP-NRS < 2 à la semaine 4 et à la semaine 16, une amélioration du score SD-NRS (Sleep Disturbance Numeric Rating Scale [échelle d'évaluation numérique des troubles du sommeil]) ≥ 4 par rapport à l'inclusion à la semaine 16, les patients avec à la fois un score EASI-75 et une amélioration du score PP-NRS ≥ 4 par rapport à l'inclusion à la semaine 16 et les patients avec à la fois un succès selon le score IGA et une amélioration du score PP-NRS ≥ 4 par rapport à l'inclusion à la semaine 16.
Caractéristiques initiales
Dans ces études, à l'inclusion, 51,0 % des patients étaient des hommes, 79,9 % étaient d'origine caucasienne et le poids moyen était de 75,0 kg. La moyenne d'âge était de 34,1 ans, 15,4 % des patients étaient des adolescents (12 à 17 ans) et 5,3 % étaient âgés de 65 ans ou plus. 70 % des patients avaient un score IGA 3 (DA modérée) à l'inclusion et 30 % des patients avaient un score IGA de 4 (DA sévère) à l'inclusion. Le score EASI moyen (ET) à l'inclusion était de 27,5 (10,5), le score PP-
NRS moyen (ET) hebdomadaire à l'inclusion était de 7,1 (1,5) (démangeaisons sévères) et le score SD-NRS moyen (ET) hebdomadaire à l'inclusion était de 5,8 (2,2). Globalement, 63,3 % des patients avaient reçu auparavant d'autres traitements systémiques pour la dermatite atopique.
Réponse clinique
Études ARCADIA 1 et ARCADIA 2 - Adultes et adolescents - période d'induction, semaine 0 à semaine 16
Le némolizumab était significativement supérieur au placebo sur le plan statistique en ce qui concerne les co-critères principaux d'évaluation cutanée à savoir le succès selon le score IGA et un score EASI-75 sur 16 semaines (Tableau 2). Les résultats pour les deux co-critères d'évaluation principaux étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥ 7).
Tableau 2 - Résultats d'efficacité du némolizumab (30 mg 1x/4 sem.) avec traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 16
|
|
ARCADIA 1 |
ARCADIA 2 |
||
|
Némolizumab + CT/ICT |
Placebo + CT/ICT |
Némolizumab + CT/ICT |
Placebo + CT/ICT |
|
|
Nombre de patients randomisés et traités (score PP-NRS à l'inclusion ≥ 4) |
620 |
321 |
522 |
265 |
|
% de patients avec un score IGA de 0 ou 1a |
35,6# |
24,6 |
37,7# |
26,0 |
|
% de patients avec un score EASI-75a |
43,5* |
29,0 |
42,1# |
30,2 |
a
Les patients qui ont reçu un traitement de secours ou dont les données étaient
manquantes ont été considérés comme non-répondeurs
*valeur de p < 0,0001,
#valeur de p < 0,001
La valeur de p ajustée pour les strates est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l'inclusion
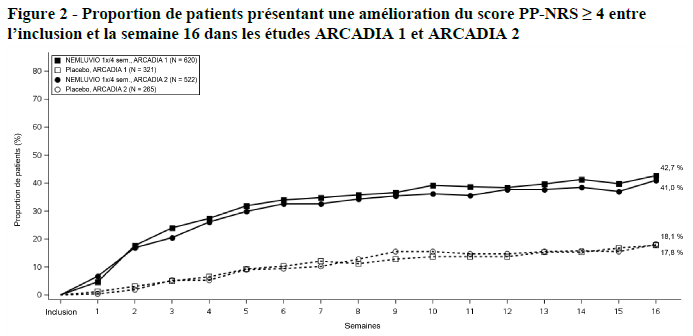
Une amélioration significative du prurit chez les patients traités par némolizumab dans les études ARCADIA 1 et ARCADIA 2 par rapport au placebo basée sur les améliorations du score PP-NRS ≥ 4 et une différence en pourcentage du score PP-NRS par rapport à l'inclusion a été observée à partir de la semaine 1 et s'est maintenue jusqu'à la semaine 16 (Tableau 3 et Figure 2). Les résultats étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥ 7).
Tableau 3 -Résultats d'efficacité sur les démangeaisons pour le némolizumab associé à un traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 jusqu'à la semaine 16
|
|
ARCADIA 1 |
ARCADIA 2 |
||
|
Némolizumab + CT/ICT |
Placebo + CT/ICT |
Némolizumab + CT/ICT |
Placebo + CT/ICT |
|
|
Nombre de patients randomisés et traités (score PP-NRS à l'inclusion ≥ 4)a |
620 |
321 |
522 |
265 |
|
% de patients présentant une améliorationdu score PP-NRS ≥ 4a |
||||
|
À la semaine 1 |
4,7§ |
1,2 |
6,7* |
0,4 |
|
À la semaine 2 |
17,7* |
3,1 |
16,9* |
1,9 |
|
À la semaine 4 |
27,4* |
6,5 |
26,1* |
5,3 |
|
À la semaine 16 |
42,7* |
17,8 |
41,0* |
18,1 |
|
% de patients avec un score PP-NRS < 2a |
|
|
|
|
|
À la semaine 4 |
16,0* |
3,7 |
15,9* |
2,6 |
|
À la semaine 16 |
30,6* |
11,2 |
28,4* |
11,3 |
|
Différence moyenne par rapport à l'inclusion (%) |
|
|
|
|
|
À la semaine 16 |
-56,1* |
-30,6 |
-55,6* |
-30,3 |
a
Les patients qui ont reçu un traitement de secours ou dont les données étaient
manquantes ont été considérés comme non-répondeurs
*valeur de p < 0,0001,
§valeur de p < 0,05
La valeur de p ajustée pour les strates est basée sur le test CMH stratifié par score PP-NRS et score IGA à l'inclusion
Chez les patients ayant un poids corporel égal ou supérieur à 90 kg, dans une analyse post-hoc de chacune des études pivots, aucune différence dans la réponse anti-inflammatoire (IGA 0 ou 1 et EASI 75) à la semaine 16 n'a été observée entre les bras némolizumab et placebo, même si l'effet a été observé au niveau de la réduction du prurit (score PP NRS).
L'échelle d'évaluation numérique des troubles du sommeil (SD-NRS) est une échelle d'évaluation quotidienne utilisée par les patients pour rapporter le degré de perte du sommeil liée à la dermatite atopique. Une amélioration significative des troubles du sommeil a été observée à la semaine 16, par rapport au placebo (Tableau 4). Les résultats étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥ 7).
Tableau 4 -Efficacité sur les troubles du sommeil pour le némolizumab associé à un traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 16
|
|
ARCADIA 1 |
ARCADIA 2 |
||
|
Némolizumab + CT/ICT |
Placebo + CT/ICT |
Némolizumab + CT/ICT |
Placebo + CT/ICT |
|
|
Nombre de patients randomisés et traités (score PP-NRS à l'inclusion ≥ 4)a |
620 |
321 |
522 |
265 |
|
% de patients présentant une améliorationdu score SD-NRS ≥ 4a Variation moyenne par rapport à l'inclusion (%) |
37,9* -64,6 |
19,9 -38,1 |
33,5* -59,7 |
16,2 -35,4 |
a
Les patients qui ont reçu un traitement de secours ou dont les données étaient
manquantes ont été considérés comme non-répondeurs
*valeur de p < 0,0001
La valeur p ajustée par strate est basée sur le test CMH stratifié par score PP-NRS et score IGA à l'inclusion
Adolescents atteints de dermatite atopique (âgés de 12 à 17 ans)
Les résultats d'efficacité des études ARCADIA 1, ARCADIA 2 à la semaine 16 pour les patients pédiatriques âgés de 12 à 17 ans sont présentés dans le Tableau 5. Les résultats dans la population de patients pédiatriques étaient dans l'ensemble cohérents avec les résultats dans la population de patients adultes. Les résultats des critères d'évaluation co-primaires et secondaires principaux étaient homogènes dans la population atteinte de prurit sévère (score PP-NRS initial ≥ 7).
Tableau 5 - Résultats d'efficacité pour le némolizumab (30 mg 1x/4 sem.) associé à un traitement concomitant par CT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 16 chez des patients pédiatriques âgés de 12 à 17 ans
|
|
ARCADIA 1 ET ARCADIA 2 |
|
|
Némolizumab + CT/ICT |
Placebo + CT/ICT |
|
|
Nombre de patients randomisés et traités (score PP-NRS à l'inclusion ≥ 4) |
179 |
90 |
|
% de patients avec un score IGA de 0 ou 1a |
48,9* |
34,4 |
|
% de patients avec un score EASI-75a |
53,4§ |
43,3 |
|
% de patients présentant une amélioration du score PP-NRS ≥ 4 a |
40,9# |
17,8 |
|
% de patients avec un score PP-NRS < 2 a |
30,1≠ |
6,7 |
|
% de patients présentant une amélioration du score SD-NRS ≥ 4 a |
31,8∞ |
20,0 |
a
Les patients qui ont reçu un traitement de secours ou dont les données étaient
manquantes ont été considérés comme non-répondeurs
≠valeur
de p < 0,0001,
# valeur de p < 0,001,
*valeur de p < 0,05,
∞valeur
de p = 0,0591,
§valeur de p = 0,1824
La valeur p ajustée par strate est basée sur le test CMH stratifié selon le score PP-NRS et le score IGA à l'inclusion
Études ARCADIA 1 et ARCADIA 2 - Adultes et adolescents - période d'entretien, semaine 16 à semaine 48
La réponse clinique chez les répondeurs au némolizumab (score IGA 0/1 ou score EASI-75 à la semaine 16) a été évaluée entre la semaine 16 et la semaine 48 dans les études ARCADIA 1 et ARCADIA 2. Pour la période de traitement d'entretien, 507 répondeurs au némolizumab ont été de nouveau randomisés pour recevoir du némolizumab 30 mg 1x/4 sem., du némolizumab 30 mg 1x/8 sem. ou un placebo 1x/4 sem. (retrait du némolizumab) associé à un traitement concomitant par CT/ICT. Les résultats d'efficacité groupés avec analyse descriptive uniquement pour cette période dans les études pivots (ARCADIA 1 et ARCADIA 2) portant sur le némolizumab à la semaine 48 sont présentés dans le Tableau 6.
Tableau 6 - Résultats d'efficacité groupés de la période d'entretien pour le némolizumab associé à un traitement concomitant parCT/ICT dans les études ARCADIA 1 et ARCADIA 2 à la semaine 48
|
|
Némolizumab + CT/ICT 1x/4 sem. N = 169 |
Némolizumab + CT/ICT 1x/8 sem. N = 169 |
Placebo + CT/ICT 1x/4 sem. (retrait du némolizumab) N = 169 |
|
% de patients avec un score IGA de 0 ou 1a |
|
|
|
|
Semaine 16 (inclusion/période d'entretien) |
84,0 |
84,0 |
77,5 |
|
Semaine 48 Différence de proportion ajustée par strate (%) IC à 95 % ajusté par strate |
61,5 11,8 (1,3, 22,3) |
60,4 10,7 (0,3, 21,0) |
49,7 |
|
% de patients avec un score EASI-75a (IC à 95 %)* |
|||
|
Semaine 16 (inclusion/période d'entretien) |
96,4 |
96,4 |
92,9 |
|
Semaine 48 Différence de proportion ajustée par strate (%) IC à 95 % ajusté par strate |
76,3 12,4 (2,7, 22,0) |
75,7 11,8 (2,1, 21,5) |
63,9 |
a Les patients qui ont reçu un traitement de secours ou dont les données étaient manquantes ont été considérés comme non-répondeurs
Efficacité et sécurité cliniques chez les adultes atteints de prurigo nodulaire
L'efficacité et la sécurité du némolizumab en monothérapie ont été évaluées dans deux études pivots randomisées, en double aveugle, contrôlées contre placebo (OLYMPIA 1 et OLYMPIA 2) dans lesquelles étaient inclus, au total, 560 patients âgés de 18 ans et plus atteints de prurigo nodulaire modéré à sévère. La sévérité de la maladie était définie par un score IGA dans l'évaluation globale des nodules de prurigo nodulaire sur une échelle de sévérité de 0 à 4. Les patients inclus dans ces deux études avaient un score IGA ≥ 3, un prurit sévère défini par une moyenne hebdomadaire du score sur l'échelle d'évaluation numérique du prurit (PP-NRS) ≥ 7 sur une échelle de 0 à 10, et 20 lésions nodulaires ou plus. Les études OLYMPIA 1 et OLYMPIA 2 évaluaient l'effet du némolizumab en monothérapie sur les signes et symptômes du prurigo nodulaire, ciblant l'amélioration des lésions cutanées et du prurit sur 16 semaines. L'étude OLYMPIA 1 comprenait une période de traitement de 24 semaines et l'étude OLYMPIA 2 une période de traitement de 16 semaines.
Dans le groupe de traitement par némolizumab, les patients pesant moins de 90 kg ont reçu des injections sous-cutanées de némolizumab 60 mg (2 injections de 30 mg) la semaine 0, suivies d'injections de 30 mg toutes les 4 semaines, et les patients pesant 90 kg ou plus ont reçu des injections sous-cutanées de némolizumab 60 mg (2 injections de 30 mg) la semaine 0 puis toutes les 4 semaines.
Critères d'évaluation
Dans les études OLYMPIA 1 et OLYMPIA 2, les deux critères d'évaluation principaux étaient identiques :
- Proportion de patients présentant une amélioration ≥ 4 par rapport à l'inclusion du score de l'échelle d'évaluation numérique du prurit maximal (PP-NRS) à la semaine 16
- Proportion de patients présentant un succès selon le score IGA (défini comme un score IGA de 0 [blanchi] ou 1 [presque blanchi] et une amélioration ≥ 2 points par rapport à l'inclusion) à la semaine 16
Les principaux critères d'évaluation secondaires comprenaient une amélioration ≥ 4 du score PP-NRS à la semaine 4 par rapport à l'inclusion, un score PP-NRS < 2 à la semaine 4 et à la semaine 16, une amélioration ≥ 4 du score l'échelle d'évaluation numérique des troubles du sommeil (SD-NRS) à la semaine 4 et à la semaine 16.
Caractéristiques initiales
Dans ces études, à l'inclusion, 59,6 % des patients étaient des femmes, 81,4 % étaient d'origine caucasienne, le poids moyen était de 82,6 kg, l'âge moyen était de 55,2 ans et 25,4 % des patients étaient âgés de plus de 65 ans. Le score PP-NRS moyen hebdomadaire à l'inclusion correspondait à une moyenne (ET) de 8,4 (0,9). Cinquante-huit (58) % des patients avaient un score IGA initial de 3 (PN modéré) et 42 % des patients avaient un score IGA initial de 4 (PN sévère).
Réponse clinique
Études pivots (OLYMPIA 1 et OLYMPIA 2) - semaine 0 à semaine 16
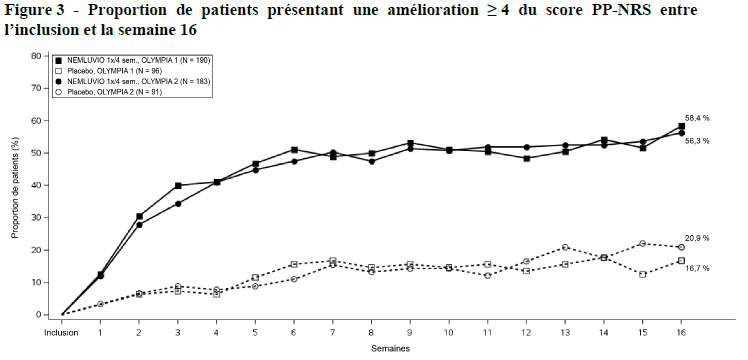
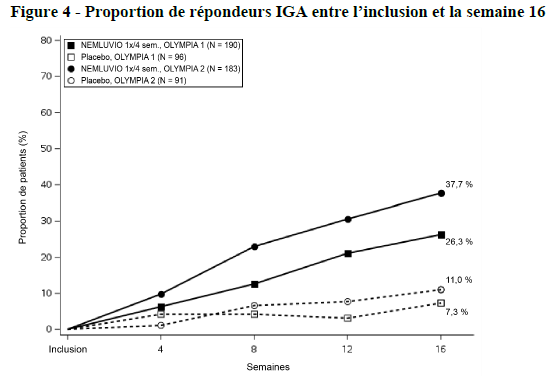
Les résultats des études pivots évaluant le traitement par némolizumab, OLYMPIA 1 et OLYMPIA 2, sont présentés dans le Tableau 7 et montrent une amélioration significative chez les patients traités par némolizumab par rapport au placebo pour les critères d'évaluation principaux (Figure 3 et Figure 4).
Tableau 7 - Résultats d'efficacité pour le némolizumab en monothérapie (1x/4 sem.) dans les études OLYMPIA 1 et OLYMPIA 2
|
|
OLYMPIA 1 |
OLYMPIA 2 |
||
|
Némolizumab |
Placebo |
Némolizumab |
Placebo |
|
|
Nombre de patients randomisés |
190 |
96 |
183 |
91 |
|
% de patients présentant une amélioration du score PP-NRS ≥ 4 par rapport à l'inclusiona |
||||
|
Semaine 4 |
41,1* |
6,3 |
41,0* |
7,7 |
|
Semaine 16 |
58,4* |
16,7 |
56,3* |
20,9 |
|
% de patients présentant un score IGA de 0 ou 1 à la semaine 16a |
26,3# |
7,3 |
37,7* |
11 |
|
% de patients avec un score PP-NRS < 2 a |
||||
|
Semaine 4 |
21,6* |
1,0 |
19,7* |
2,2 |
|
Semaine 16 |
34,2* |
4,2 |
35,0* |
7,7 |
|
% de patients présentant une amélioration du score SD-NRS ≥ 4 par rapport à l'inclusiona |
||||
|
Semaine 4 |
31,1* |
5,2 |
37,2* |
9,9 |
|
Semaine 16 |
50,0* |
11,5 |
51,9* |
20,9 |
a Si un patient recevait un traitement de secours, une stratégie de variable composite était appliquée, les données de base au moment de l'administration du traitement de secours/après le traitement de secours étaient définies comme la pire valeur possible, et la réponse était dérivée de la valeur des données de base. Les patients avec des résultats manquants étaient considérés comme des non-répondeurs.
*valeur de p < 0,0001,
#valeur de p = 0,0025 ajustée par strate à l'aide des variables de stratification randomisées (centre d'analyse et poids corporel initial (< 90 kg, ≥ 90 kg)
Absorption
Après une injection sous-cutanée initiale d'une dose de 60 mg chez des patients atteints de dermatite atopique ou de prurigo nodulaire, la concentration maximale (Cmax) moyenne estimée par la PK de population était de 6,7 (2,20) microgrammes/ml environ 6 jours après l'administration.
Après l'administration de doses multiples chez des patients atteints de dermatite atopique, les concentrations résiduelles à l'état d'équilibre estimées par la pharmacocinétique (PK) de population du némolizumab étaient de 2,63 (1,27) microgrammes/ml pour la dose de 30 mg administrée 1x/4 sem. et de 0,74 (0,44) microgrammes/ml pour la dose de 30 mg administrée 1x/8 sem.
Après l'administration de doses multiples à des patients atteints de prurigo nodulaire, la PK de population a estimé les concentrations résiduelles à l'état d'équilibre de némolizumab à 3,04 (1,23) microgrammes/ml chez des patients ayant un poids corporel < 90 kg pour 30 mg administrés 1x/4 sem. ; et à 3,66 (1,63) microgrammes/ml chez des patients ayant un poids corporel ≥ 90 kg pour 60 mg administrés 1x/4 sem.
Chez les populations atteintes de dermatite atopique et de prurigo nodulaire, les concentrations de némolizumab à l'état d'équilibre ont été atteintes à la semaine 4 après une dose de charge de 60 mg et à la semaine 12 sans dose de charge.
Une dose de charge est proposée pour les patients atteints de PN dont le poids corporel est < 90 kg. Cependant, pour les patients dont le poids corporel est ≥ 90 kg, aucune dose de charge n'est proposée, car la dose de 60 mg était suffisante pour atteindre des concentrations à l'état d'équilibre similaires à celles de la dose de 30 mg (avec une dose de charge de 60 mg) après la deuxième dose (à la semaine 8).
Distribution
D'après une analyse de la PK de population, le volume apparent de distribution (V/F) était de 7,67 l.
Biotransformation
Aucune étude spécifique du métabolisme n'a été menée car le némolizumab est une protéine. Le némolizumab devrait être métabolisé en petits peptides par les voies cataboliques.
Élimination
Le némolizumab devrait être dégradé de la même manière que les IgG endogènes. Dans l'analyse de la PK de population, la demi-vie d'élimination terminale (ET) du némolizumab a été estimée à 18,9 (4,96) jours et la clairance systémique apparente (Cl/F) a été estimée à 0,26 l/jour.
Linéarité/non-linéarité
Après une dose unique, la pharmacocinétique du némolizumab était linéaire, avec des expositions augmentant de manière proportionnelle à la dose entre 0,03 et 3 mg/kg.
Après plusieurs doses, l'exposition systémique au némolizumab a augmenté de manière approximativement proportionnelle à la dose dans la plage de doses SC allant jusqu'à 30 mg. Une légère diminution de la biodisponibilité de 9 % a été observée avec la dose SC de 60 mg.
Populations particulières
Sexe, âge et origine ethnique
Le sexe, l'âge (tranche d'âge : 12 à 85 ans pour la DA et 18 à 84 ans pour le PN) et l'origine ethnique n'ont pas eu d'effet cliniquement pertinent sur la pharmacocinétique du némolizumab.
Insuffisance hépatique
Le némolizumab, étant un anticorps monoclonal, ne devrait pas être éliminé de manière importante par voie hépatique. Aucune étude clinique n'a été réalisée pour évaluer l'effet d'une insuffisance hépatique sur la pharmacocinétique du némolizumab. Il n'a pas été constaté qu'une insuffisance hépatique légère à modérée affectait la PK du némolizumab déterminée par une analyse de la PK de population. Aucune donnée n'est disponible chez les patients atteints d'insuffisance hépatique sévère.
Insuffisance rénale
Le némolizumab, étant un anticorps monoclonal, ne devrait pas être éliminé de manière importante par voie rénale. Aucune étude clinique n'a été réalisée pour évaluer l'effet de l'insuffisance rénale sur la pharmacocinétique du némolizumab. L'analyse de la PK de population n'a pas mis en évidence d'impact cliniquement significatif d'une insuffisance rénale légère ou modérée sur l'exposition systémique au némolizumab. Les données disponibles chez les patients atteints d'insuffisance rénale sévère sont très limitées.
Poids corporel
L'exposition au némolizumab était plus faible chez les patients ayant un poids corporel plus élevé.
Dermatite atopique
La différence d'exposition systémique due au poids corporel n'a eu aucun impact cliniquement significatif sur l'efficacité. Aucun ajustement posologique en fonction du poids corporel n'est nécessaire (voir rubrique Posologie et mode d'administration).
Prurigo nodulaire
La variabilité de l'exposition systémique due au poids corporel a eu un impact cliniquement significatif sur l'efficacité sur les lésions cutanées, évaluée par la réponse IGA, mais pas sur l'amélioration du prurit, et nécessite un ajustement de la dose chez les patients atteints de PN (voir rubrique Posologie et mode d'administration).
Population pédiatrique
Dermatite atopique
Dans l'analyse de la PK de population, aucune différence cliniquement pertinente au niveau de la pharmacocinétique du némolizumab n'a été estimée chez les patients pédiatriques âgés de 12 à 17 ans par rapport aux adultes. Un ajustement de la dose dans cette population n'est pas recommandé.
Nemluvio n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité et de toxicologie en administration répétée n'ont pas révélé de risque particulier pour l'homme.
Le potentiel mutagène du némolizumab n'a pas été évalué ; cependant, on ne s'attend pas à ce que des anticorps monoclonaux altèrent l'ADN ou les chromosomes.
Aucune étude de cancérogenèse n'a été réalisée avec le némolizumab. L'évaluation des informations disponibles concernant l'inhibition de l'IL-31 et des données de toxicologie chez l'animal n'indique pas de potentiel cancérogène.
Aucun effet sur les paramètres de fertilité n'a été observé chez des singes cynomolgus sexuellement matures après un traitement par voie sous-cutanée à long terme par némolizumab. Dans le groupe des mères traitées par 25 mg/kg de némolizumab toutes les 2 semaines, du début de l'organogenèse jusqu'à la mise bas, une augmentation légère de l'incidence de décès de la progéniture a été observée au début de la période postnatale. Les expositions des mères (ASC) étaient 43 ou 34 fois plus élevées que l'exposition humaine à la dose maximale recommandée chez l'homme chez les patients atteints de DA ou de PN, respectivement. Une relation entre cette observation et le némolizumab ne peut pas être exclue.
Des instructions complètes pour l'administration de Nemluvio en stylo prérempli figurent à la fin de la notice.
Nemluvio doit être sorti du réfrigérateur pendant 30 à 45 minutes avant la reconstitution.
Inspecter à l'œil nu Nemluvio avant la reconstitution. Ne pas utiliser si la poudre n'est pas blanche, si la solution est trouble ou contient des particules. Avant l'administration, vérifier que la solution est limpide et incolore à légèrement jaune et ne contient pas de particules.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services ALLERGOLOGIE
Prescription réservée aux spécialistes et services DERMATOLOGIE
Prescription réservée aux spécialistes et services MEDECINE INTERNE
Prescription réservée aux spécialistes et services PEDIATRIE
Remboursement en fonction de l'indication (JO du 10/03/2026) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge par l'assurance maladie sont :
- traitement de la dermatite atopique modérée à sévère chez les patients âgés de 12 ans et plus qui nécessitent un traitement systémique, en cas d'échec, d'intolérance ou de contre-indication à la ciclosporine ;
- traitement des adultes atteints de prurigo nodulaire modéré à sévère qui nécessitent un traitement systémique.
Prescription réservée aux spécialistes et services ALLERGOLOGIE
Prescription réservée aux spécialistes et services DERMATOLOGIE
Prescription réservée aux spécialistes et services MEDECINE INTERNE
Prescription réservée aux spécialistes et services PEDIATRIE
Remboursement en fonction de l'indication (JO du 10/03/2026) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge par l'assurance maladie sont :
- traitement de la dermatite atopique modérée à sévère chez les patients âgés de 12 ans et plus qui nécessitent un traitement systémique, en cas d'échec, d'intolérance ou de contre-indication à la ciclosporine ;
- traitement des adultes atteints de prurigo nodulaire modéré à sévère qui nécessitent un traitement systémique.
Poudre et solvant pour solution injectable
Poudre pour solution injectable : poudre blanche lyophilisée. Solvant pour solution injectable : solution limpide, incolore.
Cartouche à usage unique en verre borosilicaté de type 1 à double compartiment dans un autoinjecteur, muni d'une aiguille en acier inoxydable.
Présentation : 1 stylo prérempli.
Chaque stylo prérempli à usage unique contient 30 mg de némolizumab par dose de 0,49 ml après reconstitution.
Le némolizumab, un anticorps monoclonal humanisé de type immunoglobuline G (IgG) modifié, est produit dans des cellules ovariennes de hamster chinois par la technologie de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Poudre pour solution injectable
Saccharose
Trométamol
Chlorhydrate de trométamol (pour ajustement du pH)
Chlorhydrate d'arginine
Poloxamère 188
Solvant
Eau pour préparations injectables
GALDERMA INTERNATIONAL
Tour Europlaza. La Défense 4
20, av André-Prothin. 92927 La Défense cdx
Tél : 01 58 86 45 45
Info médic et pharmacovigilance :
![]()
Pour commander Nemluvio :
(réservé aux pharmacies)
Tél : 04 73 69 97 57
E-mail : pharmacie@movianto.com